

AI药物化学家登Nature子刊:
原文来源:学术头条

图片来源:由无界 AI生成
药物发现是一个复杂的、多步骤的过程,其中涉及到许多化学和生物子学科的交叉领域。而人类药物化学家凭借其多年累计的专业知识在其中发挥着重要作用。
那么,人工智能(AI)能否担任药物化学家在药物发现中扮演的角色呢?答案或许是肯定的。
日前,来自诺华生物医学研究所(NIBR)和微软研究院科学智能中心(AI4Science)的研究团队,共同提出了一个机器学习模型,该模型能部分重现职业化学家在工作中积累的集体知识,这类知识通常被称为“化学直觉”。
研究团队认为,这种方法或能作为对分子建模的补充,使今后的药物研发更加高效。
相关研究论文以“Extracting medicinal chemistry intuition viapreference machine learning”为题,已发表在 Nature 子刊 Nature Communications 上。

机器学习重现药物化学家专业知识
在药物发现的“先导化合物优化”阶段,不论是湿实验室还是计算方面的药物化学家,都扮演着至关重要的角色,因为他们通常被要求确定哪些化合物需要合成和在后续优化轮次中进行评估。
为了做到这一点,药物化学家通常会审查包括活性、ADMET2 或靶标结构信息等化合物属性在内的数据。因此,一个项目的成功不仅依赖于生成的实验数据的质量,而且还依赖于从事药物化学工作团队决策的鲁棒性和合理性。
药物化学家之所以能够更高效地做出决策,是因为他们常常借助专业知识对早期药物发现的不同迭代中的成功因素具有直观的了解。
尽管以前尝试过使用基于规则的方法或简单的化学信息学可行性评分来形式化这种知识,但要捕捉到药物化学家评分中所涉及的微妙和复杂性依然是一个根本性的挑战。
出于这一动机,该研究探索了是否可以将这种专业知识提炼为机器学习模型的一部分。这样的模型可以像已经在行业中报道的其他推荐系统一样,在先导化合物优化或药物发现的其他环节中作为决策过程的辅助工具进行部署。
考虑到药物化学目前主要依赖人工工作,不可避免地受到主观偏见的影响。一些研究已经报告了药物化学家之间以及药物化学家内部评分的一致性较低。而在本研究中,研究人员希望通过借鉴多人游戏中的策略来解决一些问题。
他们将一组分子排名的任务看作是一种偏好学习问题,然后用简单的神经网络来模拟人们的个体偏好。
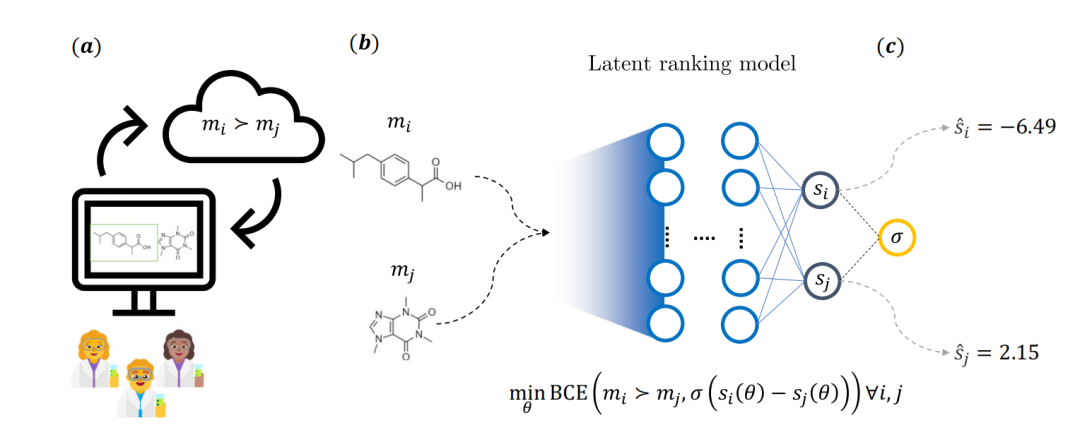
图|研究主要思路的整体示意图(来源:该论文)
具体来讲,如上图所示,分子被视为竞技比赛中的参与者,其中一方获胜的概率由化学家提供的反馈确定。为此,药物化学家要在 Web 应用程序上回答预先指定的问题提示,并选择两种分子中的一种。在此过程中,共有 35 名诺华药物化学家参与,最终共收集 5000 多个注释。
而这些反馈,催生了一个隐式得分模型。该模型采用了一种具有两个独立神经网络结构的模型,每一个分支都有固定的权重,用常见的化学信息学描述符对分子进行特征化处理。在训练期间,其参数通过二元交叉熵损失(BCE 损失)进行优化,该损失依赖于分子对的潜在得分差和化学家提供的反馈。
一旦训练完成,可以推断出任何任意分子的得分,然后可以将其用于下游化学信息学任务。
另外,该模型还可以更加准确地判断不同药物之间的相似性,该研究提出的学习评分函数比传统的药物相似性评估指标(QED)更加精准。
值得注意的是,为了促进研究的可重复性和该领域的进一步发展,研究人员还提供了一个名为“MolSkill”的软件包,其中包含了该模型和匿名响应数据。
机器学习在药物化学中的不足与应用
然而,尽管该模型可以重现药物化学家在工作中积累的知识,但也存在一些局限性。首先,为捕捉化学直觉,数据收集过程中所提出的问题一直都很模糊。
另外,虽然提出的研究设计导致与以前的研究相比参与者之间的一致性更高,但成对比较方法也并不是完美的。
此外,“Flatland谬论”使得人类往往倾向于将高维问题简化为一小组可以认知追踪的变量,而这种简化可能受每个药物化学家特点的影响。
然而,研究团队表示,本次研究提出的模型不仅限于当前研究的应用范围。具体来说,讨论的框架可以扩展到药物发现领域的其他可量化但却昂贵的可观测值。此外,它可以为化学空间中尚未被探索的领域提供见解。
免责声明:数字资产交易涉及重大风险,本资料不应作为投资决策依据,亦不应被解释为从事投资交易的建议。请确保充分了解所涉及的风险并谨慎投资。OKEx学院仅提供信息参考,不构成任何投资建议,用户一切投资行为与本站无关。

和全球数字资产投资者交流讨论
扫码加入OKEx社群
industry-frontier

